


Aιμορραγική Τελαγγειεκτασία: Η νόσος Osler-Weber-Rendu (OWRD) είναι μια σπάνια αυτοσωμική επικρατούσα διαταραχή που επηρεάζει τα αιμοφόρα αγγεία σε όλο το σώμα (προκαλώντας αγγειακή δυσπλασία) και έχει ως αποτέλεσμα την τάση για αιμορραγία.
Η νόσος Osler-Weber-Rendu (OWRD) είναι μια σπάνια αυτοσωμική επικρατούσα διαταραχή που επηρεάζει τα αιμοφόρα αγγεία σε όλο το σώμα (προκαλώντας αγγειακή δυσπλασία) και έχει ως αποτέλεσμα την τάση για αιμορραγία. Η κατάσταση είναι επίσης γνωστή ως κληρονομική αιμορραγική τελαγγειεκτασία [HHT] οι δύο όροι χρησιμοποιούνται εναλλακτικά σε αυτό το άρθρο.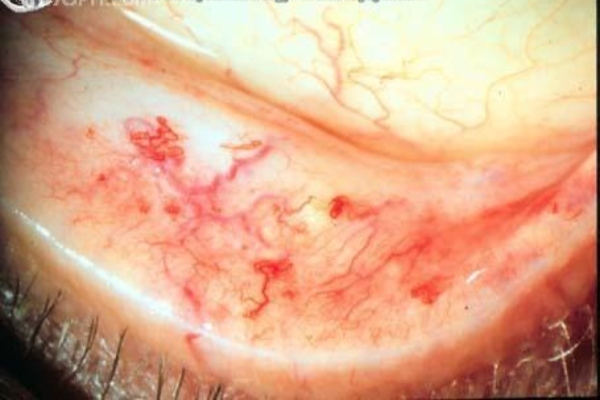 Η πρόγνωση ποικίλλει, ανάλογα με τη σοβαρότητα των συμπτωμάτων. γενικά, είναι καλό, εφόσον η αιμορραγία αναγνωρίζεται έγκαιρα και ελέγχεται επαρκώς. Η HHT εκδηλώνεται με βλεννογονοδερματικές τελαγγειεκτάσες και αρτηριοφλεβικές δυσπλασίες (AVMs), μια πιθανή πηγή σοβαρής νοσηρότητας και θνησιμότητας. Οι βλάβες μπορεί να επηρεάσουν τον ρινοφάρυγγα, το κεντρικό νευρικό σύστημα (ΚΝΣ), τον πνεύμονα, το ήπαρ και τον σπλήνα, καθώς και το ουροποιητικό σύστημα, το γαστρεντερικό (GI), τον επιπεφυκότα, τον κορμό, τα χέρια και τα δάχτυλα.
Η πρόγνωση ποικίλλει, ανάλογα με τη σοβαρότητα των συμπτωμάτων. γενικά, είναι καλό, εφόσον η αιμορραγία αναγνωρίζεται έγκαιρα και ελέγχεται επαρκώς. Η HHT εκδηλώνεται με βλεννογονοδερματικές τελαγγειεκτάσες και αρτηριοφλεβικές δυσπλασίες (AVMs), μια πιθανή πηγή σοβαρής νοσηρότητας και θνησιμότητας. Οι βλάβες μπορεί να επηρεάσουν τον ρινοφάρυγγα, το κεντρικό νευρικό σύστημα (ΚΝΣ), τον πνεύμονα, το ήπαρ και τον σπλήνα, καθώς και το ουροποιητικό σύστημα, το γαστρεντερικό (GI), τον επιπεφυκότα, τον κορμό, τα χέρια και τα δάχτυλα.
Υπάρχουν 2 κύριοι τύποι HHT που προκαλούνται και οι δύο από ετερόζυγες μεταλλάξεις. Η HHT1 περιλαμβάνει μια μετάλλαξη στην ενδογλίνη (ENG). Με αυτόν τον τύπο, οι ασθενείς, ιδιαίτερα οι γυναίκες, διατρέχουν υψηλότερο κίνδυνο να αποκτήσουν πνευμονικά και εγκεφαλικά AVM. Το HHT2 περιλαμβάνει μια μετάλλαξη στον υποδοχέα τύπου 1 της ακτιβίνης Α (ACVRL1), επίσης γνωστή ως ALK1. Οι ασθενείς με HHT2 έχουν υψηλότερο κίνδυνο να αποκτήσουν ηπατικά AVM. Το ENG περιλαμβάνει περίπου το 61% και το ACVRL1 περιλαμβάνει περίπου το 37% των μεταλλάξεων που είναι γνωστό ότι προκαλούν HHT. Έχουν βρεθεί μεταλλάξεις στον παράγοντα διαφοροποίησης ανάπτυξης 2 (GDF2). Αυτά κωδικοποιούν την πρωτεΐνη που συνδέεται με την ενδογλίνη και το ACVRL1. Τέλος, υπάρχουν μεταλλάξεις στο SMAD4 που κωδικοποιεί μια πρωτεΐνη που μεταδίδει σήματα από τον υποδοχέα του μετασχηματιστικού αυξητικού παράγοντα-βήτα (TGF-beta). Αυτή η μετάλλαξη περιλαμβάνει μόνο περίπου το 2% των περιπτώσεων. Οι ασθενείς με αυτή τη γονιδιακή μετάλλαξη εμφανίζουν νεανική γαστρεντερική πολυποδίαση και HHT.
Ο επιπολασμός της HHT είναι 1,5 έως 2 άτομα/10.000. Ωστόσο, ορισμένες πηγές λένε ότι είναι υψηλότερο λόγω της μεταβλητής διείσδυσης και επειδή τα συμπτώματα δεν εμφανίζονται παρά αργότερα στην ενήλικη ζωή. Η HHT έχει υψηλότερο επιπολασμό σε ορισμένους πληθυσμούς, όπως οι κάτοικοι της Αφρο-Καραϊβικής του Κουρασάο και του Μποναίρ. Οι μεταλλάξεις που εντοπίζονται στο HHT διαταράσσουν τις οδούς που προκαλούνται από τον TGF-βήτα στα αγγειακά ενδοθηλιακά κύτταρα. Η διαταραχή σε αυτό το μονοπάτι έχει ως αποτέλεσμα την ανώμαλη ανάπτυξη των αιμοφόρων αγγείων που οδηγεί σε εξαιρετική ευθραυστότητα και αρτηριοφλεβικές δυσπλασίες. Η ιστοπαθολογική περιγραφή των τηλαγγειεκτασιών χαρακτηρίζεται ότι έχει διεσταλμένα τριχοειδή αγγεία επενδεδυμένα με επίπεδα ενδοθηλιακά κύτταρα. Τα AVM χαρακτηρίζονται ότι έχουν ένα μείγμα αγγείων με παχύ και λεπτό τοίχωμα στο χόριο.
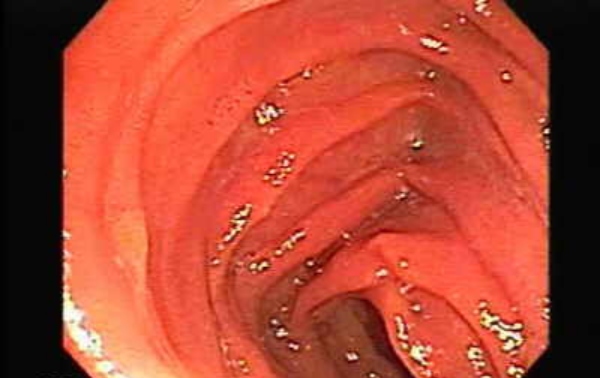
Η κληρονομική αιμορραγική τελαγγειεκτασία (HHT) έχει ως αποτέλεσμα σημαντική δυσφορία στην καθημερινή ζωή των ασθενών και των οικογενειών τους. Αυτή η ασθένεια θέτει τους ασθενείς σε κίνδυνο για επιπλοκές που απειλούν τη ζωή τους. Είναι κρίσιμο για αυτούς τους ασθενείς να κάνουν τον κατάλληλο διαγνωστικό έλεγχο που χρειάζονται για την πρόληψη της εσωτερικής αιμορραγίας ή ακόμα και του θανάτου και να έχουν μια διεπαγγελματική ομάδα κλινικών ιατρών και ειδικευμένων νοσοκόμων που θα βοηθούν στο συντονισμό της μετέπειτα φροντίδας. Αυτό θα έχει ως αποτέλεσμα την καλύτερη έκβαση του ασθενούς

Ακολουθήστε το healthweb.gr στο Google News και μάθετε πρώτοι όλες τις ειδήσεις
Ακολουθήστε το healthweb.gr στο κανάλι μας στο YouTube













